
科研服务
立足于生命科学,为基础研究领域科学工作者提供生物学高端技术服务
瑞源生物
相关产品

- 产品描述
-
服务描述
真核有参转录组核心是以已注释的完整真核生物参考基因组为“坐标模板”,通过高通量测序(如 RNA-seq)捕获细胞内的全部转录本,再通过序列比对、注释、定量等生物信息学分析,解析转录本的结构、表达水平及调控规律。可以解决转录图谱的绘制、基因差异表达模式及进化分析等各方面研究问题,已经被广泛应用于临床诊断和药物研发等诸多领域。
应用领域
农林领域:抗逆胁迫机制,生长发育机制,育种保护研究等。
微生物领域:致病机理,耐药机制,病原体-宿主相互作用研究等。
海洋水产:渔业资源,海水养殖,渔业环境与水产品安全等。
基础医学、临床诊断:生物标志物,疾病机理机制,疾病分型,个性化治疗等。
技术优势
样本处理经验丰富:各类动植物组织和细胞、微生物、外泌体等。
平台优势:llumina/T7双平台灵活可选、质控严格、数据质量高。
分析内容丰富:分析丰富、可进行多种组学联合分析。
服务流程

生物信息学分析流程
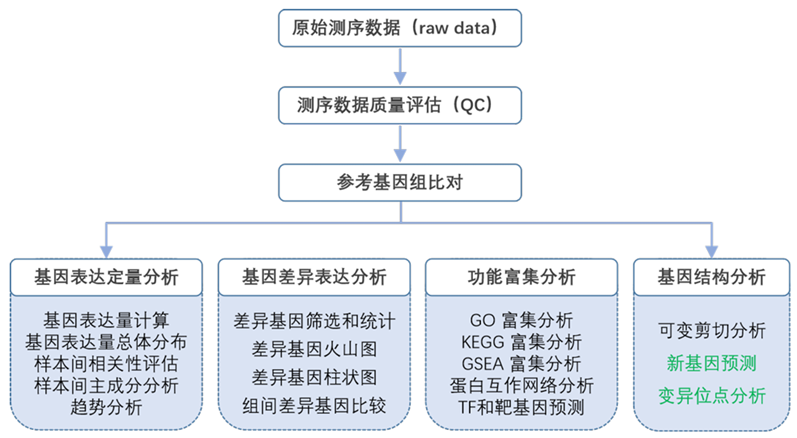
备注:标准报告包含所有黑色字体分析内容,不包含绿色字体分析内容
样品接收标准
样本
要求
备注 新鲜植物组织
≥400 mg ,≥3个生物学重复
液氮速冻,-80℃保存,干冰运输,1天(5kg)2-3天(10-20kg)3-5天(20-30kg)
新鲜动物组织
≥200 mg,≥3个生物学重复
新鲜培养细胞
≥8x107 个,≥3个生物学重复
新鲜采集全血
≥4 mL,≥3个生物学重复
菌体
≥400 mg,≥3个生物学重复
服务流程、周期及交付
服务流程 工作日 交付材料 RNA提取及质检 28 1、原始数据、结果图片
2、标准实验报告(含实验步骤、流程)
3、生信分析:基因表达定量分析、基因差异表达分析、功能富集分析、基因结构分析
文库构建及质控 上机测序 生信分析 实验报告 数据分析
服务标准 分析内容 拟解决问题 标准数据分析
质控 获得高质量数据 参考基因组比对(有参)/denovo组装(无参) 获得后续分析的基因 新基因预测 发现新基因 基因表达水平 获得基因的表达丰度 表达差异分析 鉴定差异基因表达情况 功能注释与富集 确定基因的功能特征及代谢通路 蛋白互作网络分析 利用网络图挖掘核心基因 转录因子分析 对差异基因进行转录因子注释 可变剪切分析 挖掘基因的不同剪切类型 高级数据分析
WGCNA 获得与表型强关联的核心基因 GSEA分析 检验目标基因间的富集情况 趋势聚类分析 探讨基因的表达趋势模式 基因融合分析 探索基因的嵌合情况 案例报告
(1)基因表达水平分析
基因表达水平计算
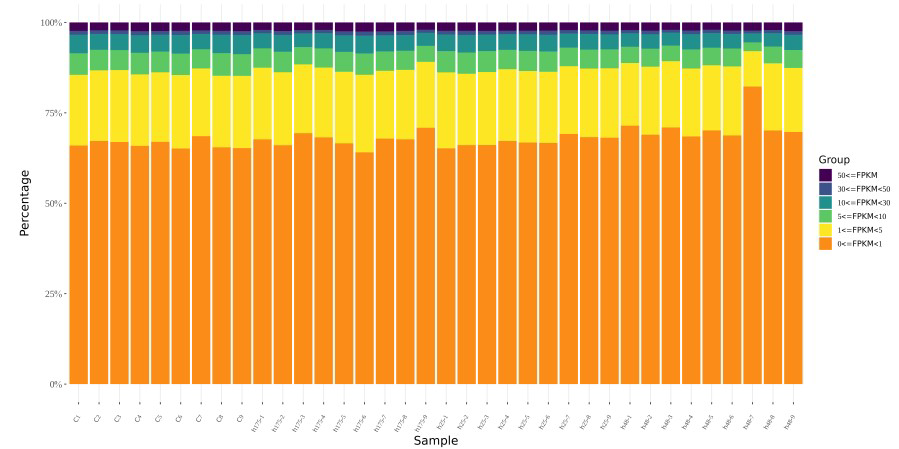
图:不同表达水平区间的基因数量统计图
(2)样本基因表达量总体分布
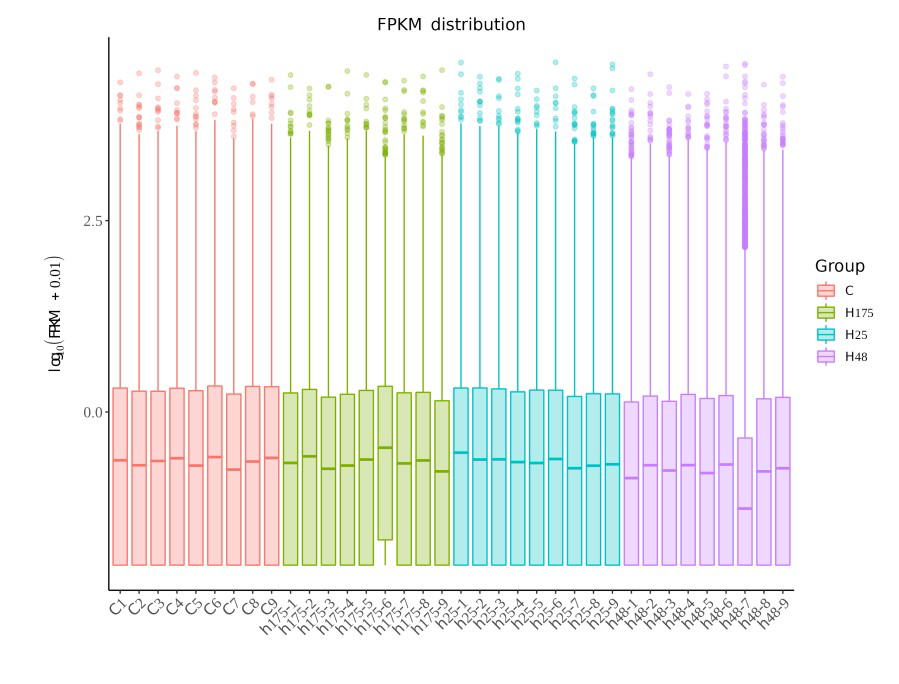
图:各样本FPKM箱线图
(3)RNA-seq 整体质量评估
样本间相关性评估
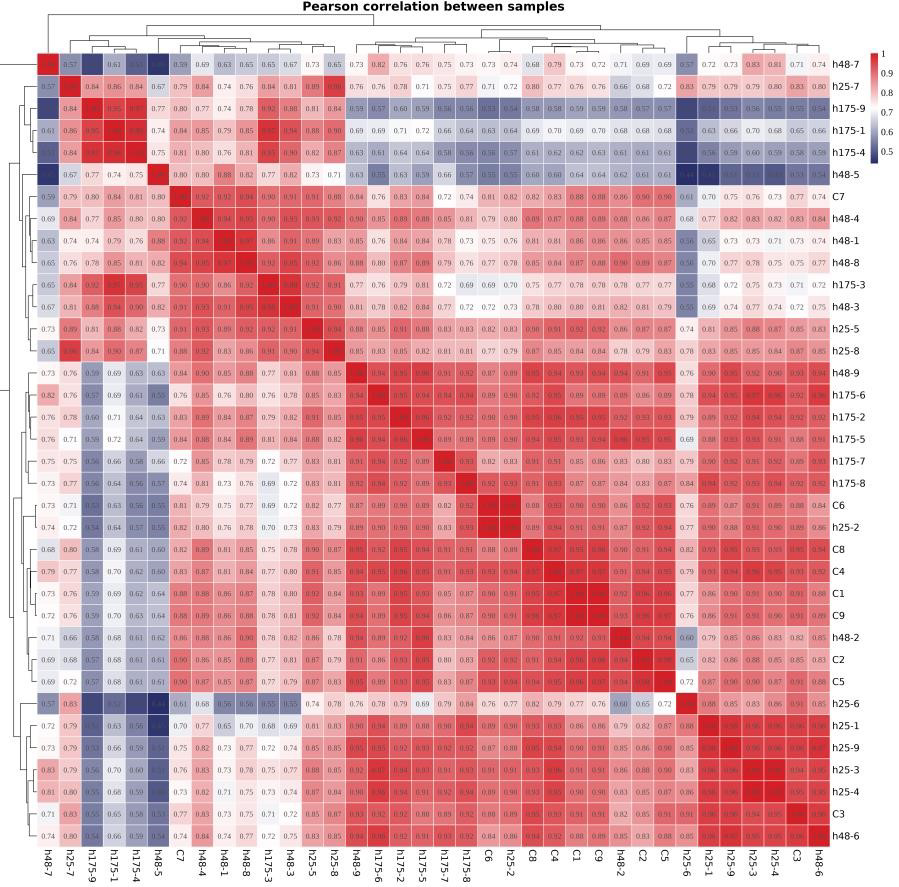
图:样本间相关系数热图
(4)差异表达分析
筛选差异基因
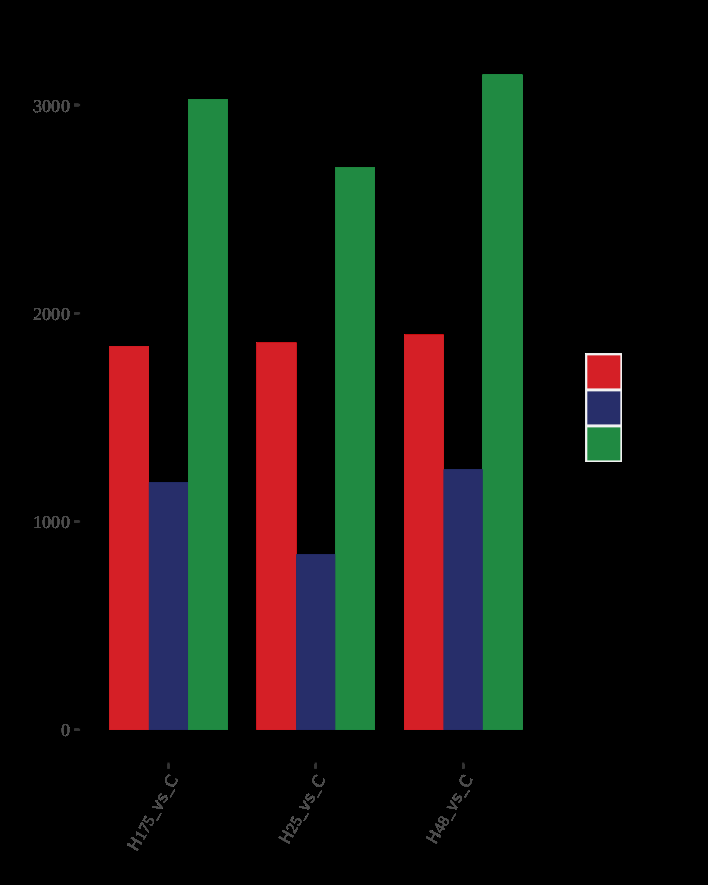
图:差异表达基因统计柱状图
(5)差异表达基因功能注释和富集分析
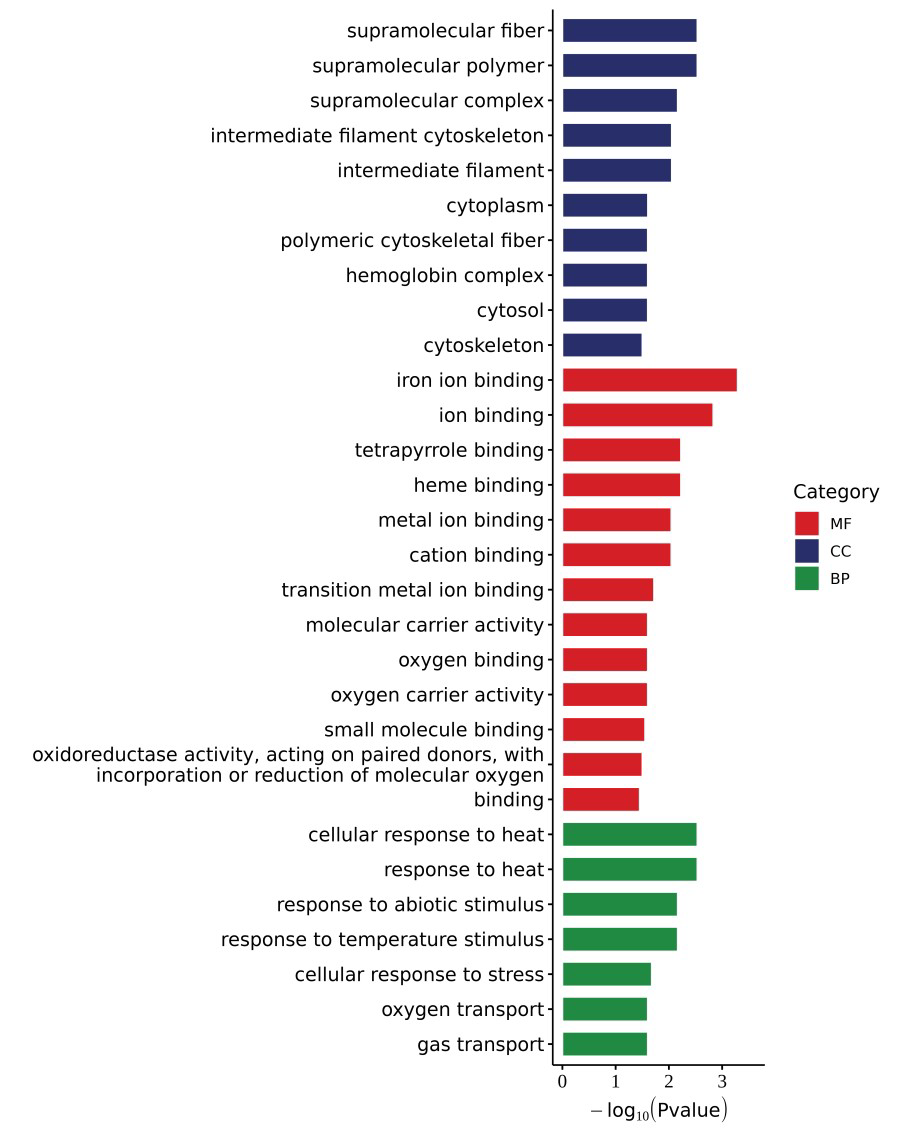
图:差异基因 GO 富集柱状图
(6)差异基因蛋白互作网络分析
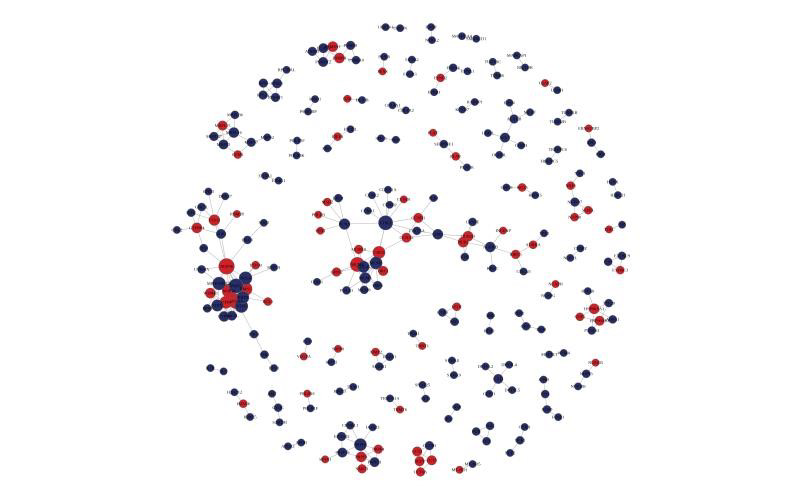
图:差异表达基因的互作网络图(top300)
(7)转录因子分析
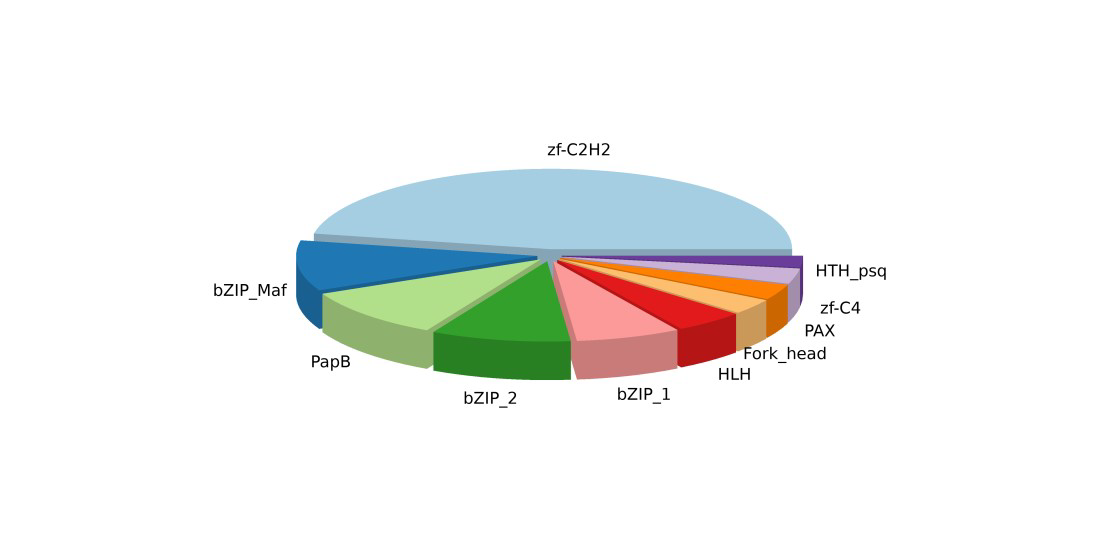
(8)可变剪切分析
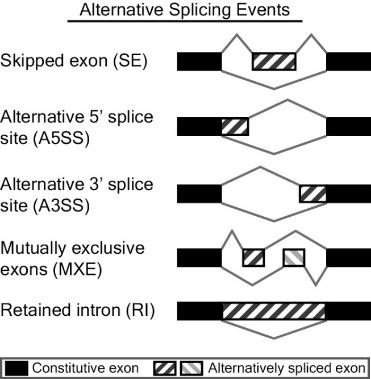
图:可变剪切事件分类
参考文献
[1]Marcel Martin. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. (cutadapt).
[2] Daehwan Kim, Ben Langmead, Steven L Salzberg. (2015). HISAT: a fast spliced aligner with low memory requirements. Nature methods. (HISAT2).
[3] Simon Anders, Paul Theodor Pyl, Wolfgang Huber. (2014). HTSeq-A Python framework to work with highthroughput sequencing data. Bioinformatics. (HTSeq).
[4] Love, M.I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. (DESeq2).
[5] Zuguang Gu, Roland Eils , Matthias Schlesner. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. (ComplexHeatmap).
[6] Robinson M. D., McCarthy D. J. & Smyth G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. (edgeR).
[7] Kanehisa M., M. Araki, et al. (2008). KEGG for linking genomes to life and the environment. Nucleic acids research. (KEGG).
[8] Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. [2003]. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research.
[9] Shen S., Park JW., Lu ZX., Lin L., Henry MD., Wu YN., Zhou Q., Xing Y. [2014]. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. Proc Natl Acad Sci USA. (rMATS).
[10] Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT& Salzberg SL. [2015]. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology. (Stringtie).
声明:本公司出售产品只能用于科研目的,不得用于诊断或者治疗!

