
科研服务
立足于生命科学,为基础研究领域科学工作者提供生物学高端技术服务
瑞源生物
相关产品

- 产品描述
-
服务简介
南京瑞源生物采用先进的Illumina测序平台,快速、高效地读取高质量的测序数据,以精准快捷为核心,准确检测样品间的重要变异信息,对候选基因进行注释后能通过荧光定量PCR、EMSA、酵母单杂、酵母双杂等实验进行候选基因验证。仅需客户提供原始材料,南京瑞源生物进行建库测序、BSA分析、基因挖掘、功能验证一条龙服务,节约客户等待时间。
技术原理
BSA(Bulked segregation analysis)即混合分组分析(BSA,or 集群分离分析法),也称分离群体分组分析。是指利用目标性状存在极端表型差异的两个亲本构建分离群体,在子代分离群体中,选取两组表型差异极端的个体分别构建混合池,结合高通量测序技术对混合样本测序,比较两组群体在多态位(SNP)的等位基因频率(AF)是否具有显著差异,定位与目标性状相关联的位点并对其进行注释,研究控制目标性状的基因及其分子机制。
应用领域
瑞源生物重测序BSA分析主要应用于定位植物、动物中控制重要性状的基因,如产量、抗病性、耐逆性等相关基因,用于辅助育种;
1、基因定位:识别与特定表型相关的基因或基因组区域;
2、候选基因挖掘:发现可能导致表型变异的候选基因;
3、分子标记开发:开发与目标性状关联的分子标记,用于辅助选择或育种;
4、基因功能研究:通过关联分析结果,预测基因功能并设计后续实验进行验证。
技术优势
1、创新技术:采用先进的Illumina测序平台,快速、高效地读取高质量的测序数据;主要使用BWA[1]软件进行短序列与参考基因组比对,GATK[2]软件实现变异检测,用SnpEff[3]软件进行变异注释
2、效率提升:采用ΔSNP Index及ED两种方法定位SNP
3、候选基因功能验证:南京瑞源生物具有多年的候选基因功能验证经验,通过荧光定量PCR、EMSA、酵母单杂、酵母双杂等实验,批量化的流水线作业减少个体实验的误操作差异概率;
4、节约客户时间:仅需客户提供原始材料,南京瑞源生物进行建库测序、BSA分析、基因挖掘、功能验证一条龙服务。
服务流程
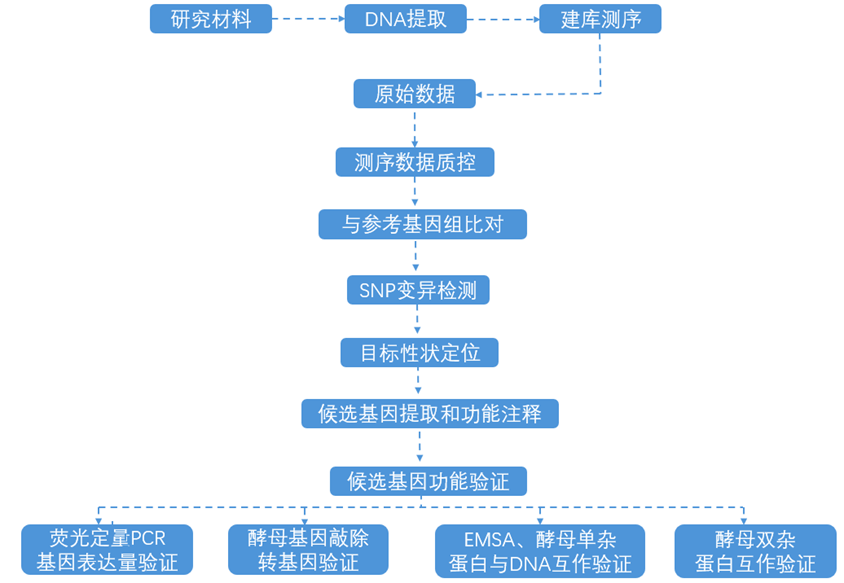
收样指南
样本类型 送样量 备注 基因组DNA > 0.5 μg 浓度>20 ng/uL,体积>15 μL,主条带完整,
OD260/280在1.7-1.9之间,-20℃保存,干冰运输
植物组织 鲜重>0.5g 液氮速冻,-80℃保存,干冰运输 动物组织 鲜重>0.2g 新鲜组织,液氮速冻,-80℃保存。干冰运输 细胞 细胞量>1x106 细胞沉淀,液氮速冻,-80℃保存,干冰运输 血液(抗凝) 全血>1ml 液氮速冻,-80℃保存,干冰运输 亲本选择表型差异极端的两个,子代选择极端表型个体各20各。建议每个子代样本单独提取 DNA 后再等量混合,可以减少背景噪音,避免系统误差;需要选择有参考基因组的物种。
服务内容
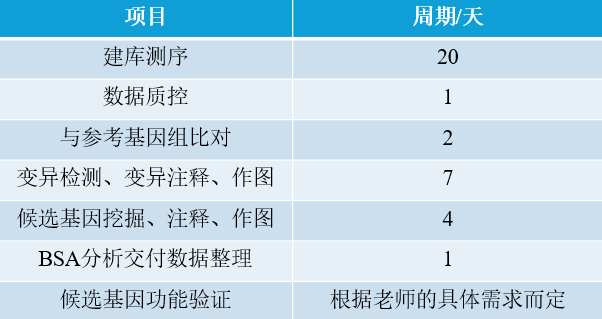
服务说明
1、BSA性状定位适用于对家系群体的单个性状进行主效定位;建议尽可能选择性状差异单⼀,杂合位点少的亲本。对于⽬标性状亲本,有多种⽅法获得,例如 EMS 诱变,⾃然个体,紫外线诱变等。
2、如果研究的性状是EMS诱导的突变性状,可以只测两个⼦代池(野⽣型+突变型),或者测 1个亲本(野⽣型)和1个⼦代池(突变型);如果研究的性状是数量性状,建议测2个亲本和2个⼦代池;
3、建议每个子代样本单独提取 DNA 后再等量混合,可以减少背景噪音,避免系统误差。
4、对于无参考基因组的物种、高度杂合的群体,目前无法进行BSA分析
案例展示
BSA分析:
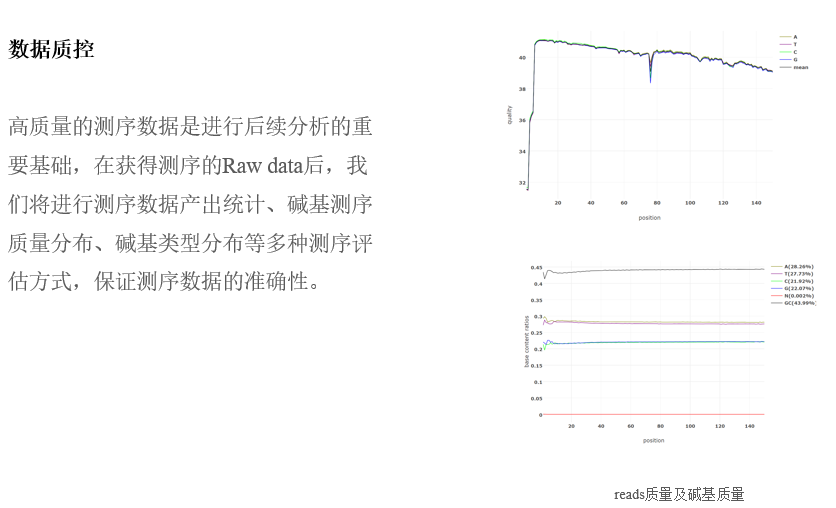
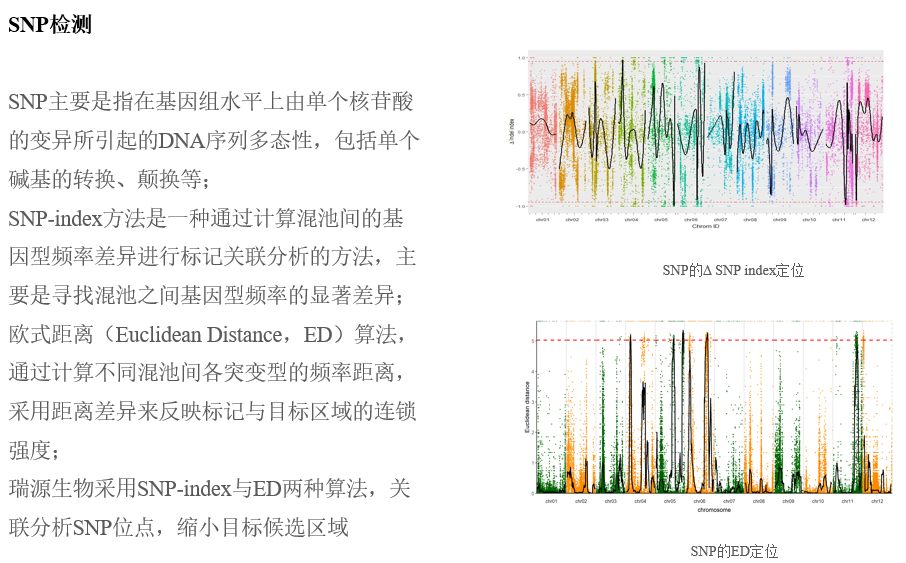
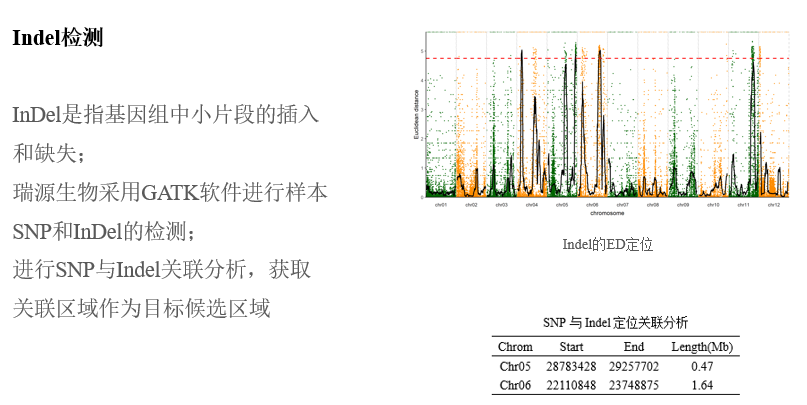

候选基因验证:
BSA分析中,除了获得候选区域,候选基因的功能验证也十分重要,需要利用实验证明候选基因与性状表型间的联系。可根据客户产品需求,南京瑞源生物提供以下候选基因验证服务:
1、荧光定量PCR
2、EMSA
3、酵母单杂互作验证
4、酵母双杂互作验证
实验材料
1、测序平台:Illumina高通量测序平台。
2、实验方案设计:从材料选取,建库测序,到数据分析,每一步都会进行科学、缜密的设计,以保障高质量研究成果。
3、支持的分析方法:ED算法和SNP-index算法。ED法具有很强的去除背景噪音能力,一大优势就是无需亲本的信息即可进行定位;SNP-index方法通常需要亲本的测序信息,可排除两个亲本相对于参考基因组共有的SNP,去除背景噪音,也可去除一部分SNP index趋近于1但是与目标性状并非连锁的标记。
4、支持的候选基因验证方法:荧光定量PCR、EMSA、酵母单杂、酵母双杂等实验,具体实验根据客户需求进行安排。
参考文献
[1] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009 Jul 15;25(14):1754-60. doi: 10.1093/bioinformatics/btp324. Epub 2009 May 18. PMID: 19451168; PMCID: PMC2705234.
[2] McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010 Sep;20(9):1297-303. doi: 10.1101/gr.107524.110. Epub 2010 Jul 19. PMID: 20644199; PMCID: PMC2928508.
[3] Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012 Apr-Jun;6(2):80-92. doi: 10.4161/fly.19695. PMID: 22728672; PMCID: PMC3679285.
[4] Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol. 2012 Jan 22;30(2):174-8. doi: 10.1038/nbt.2095. PMID: 22267009.
[5] Hill JT, Demarest BL, Bisgrove BW, Gorsi B, Su YC, Yost HJ. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013 Apr;23(4):687-97. doi: 10.1101/gr.146936.112. Epub 2013 Jan 8. PMID: 23299975; PMCID: PMC3613585.
声明:本公司出售产品只能用于科研目的,不得用于诊断或者治疗!

