
科研服务
立足于生命科学,为基础研究领域科学工作者提供生物学高端技术服务
瑞源生物
相关产品

- 产品描述
-
服务描述
蛋白质-蛋白质相互作用(Protein Protein Interaction,PPI)是指蛋白质之间通过物理接触或化学连接进行相互作用的现象。这种相互作用在细胞的生理活动中起着至关重要的作用,如信号传导、代谢路径、基因表达调控等。传统的PPI预测方法依赖于实验手段,如酵母双杂交(Y2H)和免疫共沉淀(Co-IP),这些方法尽管准确性高,但通常耗时、成本高且适用范围有限。为了应对这一挑战,计算生物学家开发了基于计算方法的PPI预测工具,这些工具可以根据蛋白质的序列或结构信息预测可能的相互作用。
应用领域
疾病的发生机制、信号传导机制、蛋白质功能预测等方面。
技术优势
高精度筛选:新升级四轮筛选,引入最新AlphaFold3互作分析软件,输出高置信度互作结果
高通量初筛:储备100+物种的全基因组数字文库,20+物种的转录因子文库,可直接开展筛选,高通量筛选上万个蛋白,节约等待时间
高质量图片:精品版服务提供五组蛋白三维模型结合位点分析图,清晰展示结合位点、氢键及距离信息,提供全面的互作细节
服务流程

样品接收标准
样本
要求
备注
基因序列
需提供CDS文件
若数据库版本要求请说明
服务流程、周期及交付
精品版
服务流程
工作日
交付材料
数字文库构建
15个
1、三轮筛选全数据Excel表(包括基因编号、建模评分、是否互作)
2、五对结合位点分析图
诱饵基因建模分析
1个
DOCK初筛对接评估
2个
大模型互作深度预测分析
5个
精细互作分析
5个
数据整理和分析
1个
普通版
服务流程
工作日
交付材料
数字文库构建
15个
两轮数字文库筛选全数据Excel表(包括基因编号、建模评分、是否互作)
诱饵基因建模分析
1个
DOCK初筛对接评估
2个
大模型互作深度预测分析
5个
数据整理和分析
2个
注:普通版为两轮筛选,无AlphaPulldown、AlphaFold3筛选和结合位点析。
案例展示
1、bait.pdb为诱饵基因的蛋白三维结构。

图:bait蛋白的三维结构
2、如图所示为第一轮预测得到的结果:其中A列为诱饵基因的ID;B列为诱饵基因的蛋白序列;C列为诱饵基因的三维结构建模分值;D列为文库中基因的名称;E列为文库基因的蛋白序列;F为文库中基因的三维结构建模分值;G列为文库中基因的功能注释;H列为Megadock对接的最后得分。
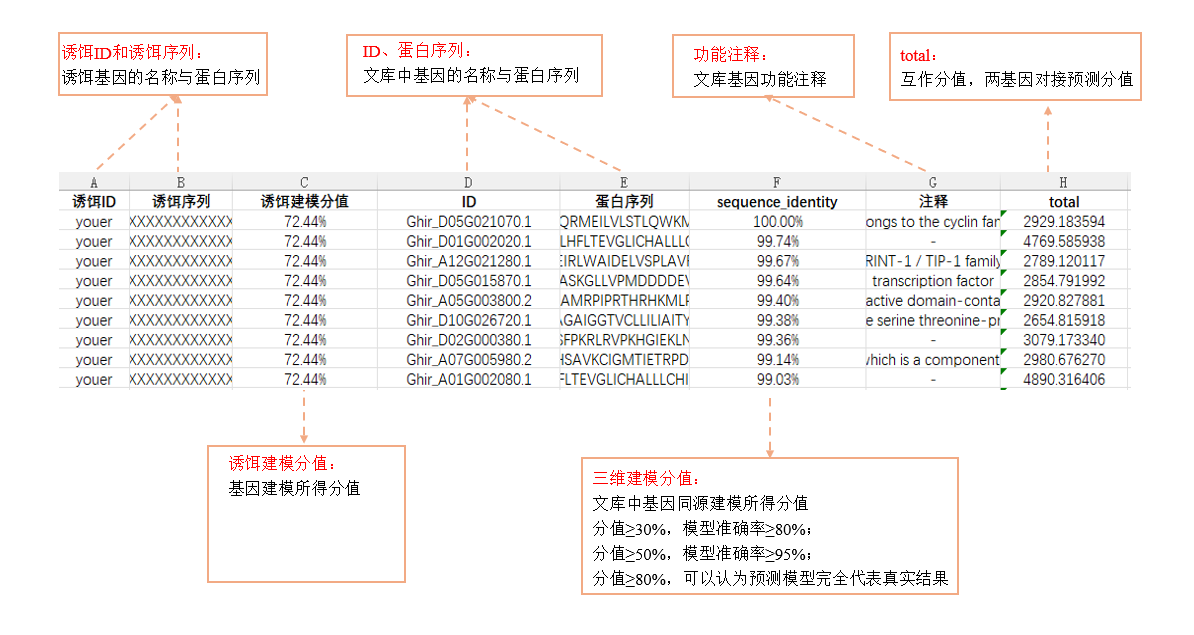
图:第一轮筛选结果(Megadock初筛)
3、提取第一轮预测结果中所有的对接数据特征,使用自主研发的AI-PPI模型进行精筛,如图所示为第二轮预测的结果,在最后一列中标注了两个蛋白是否互作,其中1代表互作,0代表不互作。
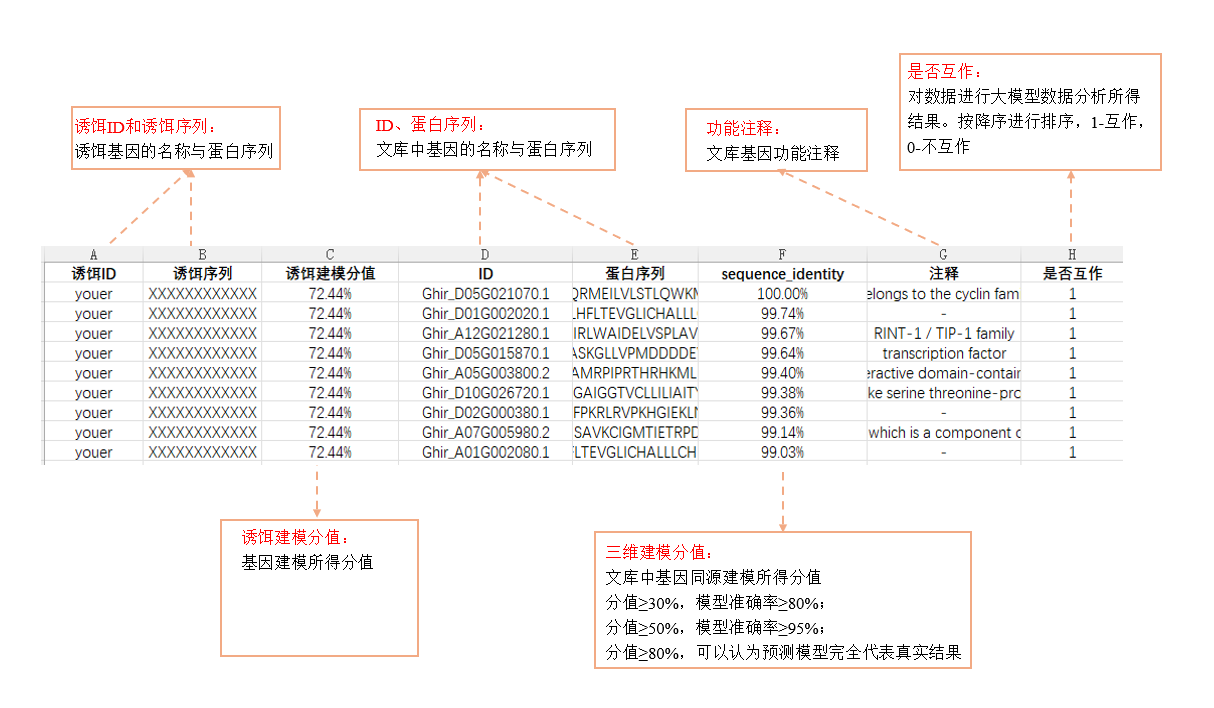
图:第二轮筛选结果(自研AI-PPI模型精筛)
4、第三轮使用AlphaPulldown程序对可能为假阳性的预测结果以及想要重点关注的蛋白互作对详细分析,结果如图所示。
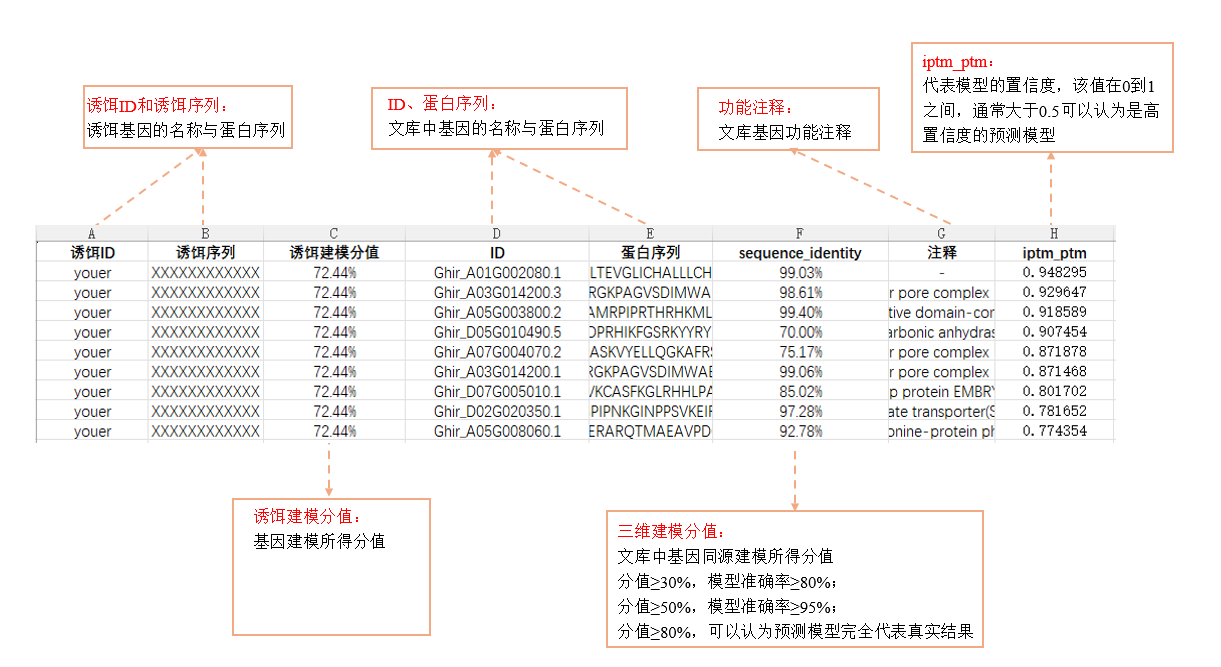
图:第三轮筛选结果(AlphaPulldown,精品版)
5、最后选取 AlphaPulldown 综合打分排名前10的蛋白对,进入AlphaFold3的一对一精细建模与再评估。综合ipTM(界面置信度)与pTM(整体折叠置信度)形成联合评分,iptm+ptm值越大,代表预测的可靠性越高,一般情况下iptm+ptm > 0.75认为有较好的互作可能性。
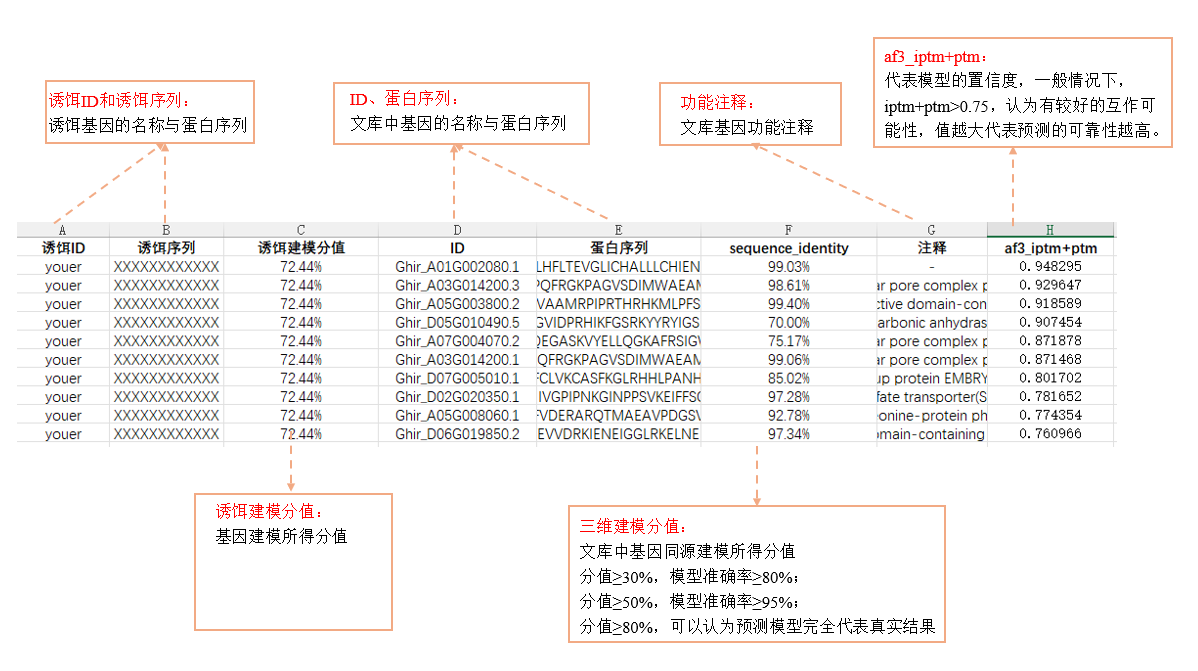
图:第四轮筛选结果(AlphaFold3,新升级10对预测,精品版)
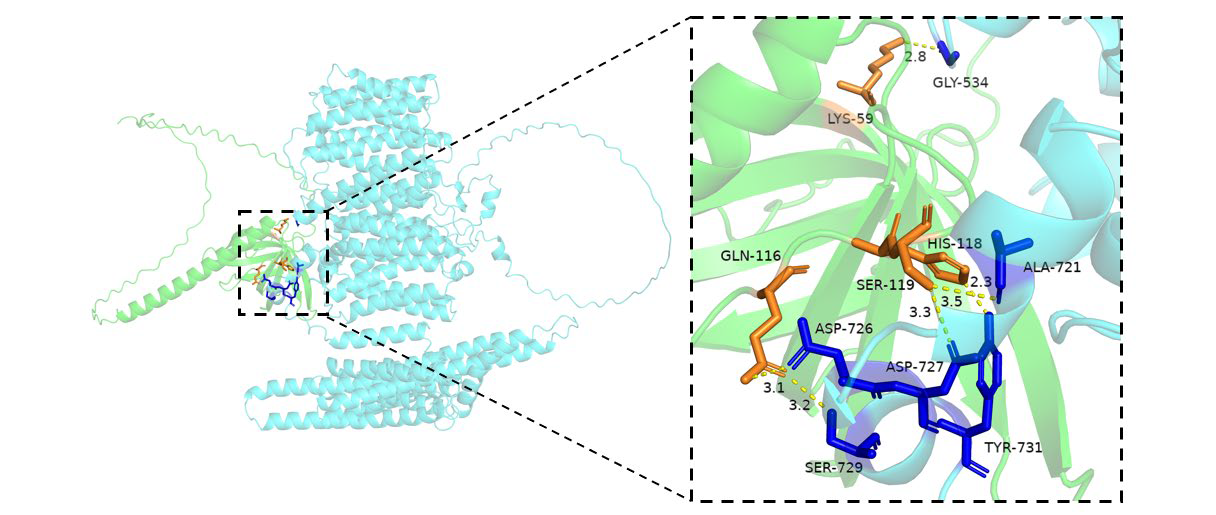
图:bait蛋白与筛选蛋白关键残基互作图(精品版)
参考文献
[1] Rao V S, Srinivas K, Sujini G, et al. Protein‐protein interaction detection: methods and analysis[J]. International journal of proteomics, 2014, 2014(1): 147648.
[2] Sudha G, Nussinov R, Srinivasan N. An overview of recent advances in structural bioinformatics of protein–protein interactions and a guide to their principles[J]. Progress in biophysics molecular biology, 2014, 116(2-3): 141-150.
[3] Ohue M, Matsuzaki Y, Uchikoga N, et al. MEGADOCK: an all-to-all protein-protein interaction prediction system using tertiary structure data[J]. Protein peptide letters, 2014, 21(8): 766-778.
[4] Yu D, Chojnowski G, Rosenthal M, et al. AlphaPulldown—a python package for protein–protein interaction screens using AlphaFold-Multimer[J]. Bioinformatics, 2023, 39(1): btac749.
[5] Schwede T, Kopp J, Guex N, et al. SWISS-MODEL: an automated protein homology-modeling server[J]. Nucleic acids research, 2003, 31(13): 3381-3385.
[6] Oughtred R, Stark C, Breitkreutz B-J, et al. The BioGRID interaction database: 2019 update[J]. Nucleic acids research, 2019, 47(D1): D529-D541.
[7] Mering C V, Huynen M, Jaeggi D, et al. STRING: a database of predicted functional associations between proteins[J]. Nucleic acids research, 2003, 31(1): 258-261.
[8] Evans R, O’neill M, Pritzel A, et al. Protein complex prediction with AlphaFold-Multimer[J]. biorxiv, 2021: 2021.10. 04.463034.
[9] Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
声明:本公司出售产品只能用于科研目的,不得用于诊断或者治疗!

